Once you have counted cells in each of the squares, you perform the hemocytometer calculations based on your total counts, dilution factor, initial volume and desired final density.
Now, here’s what you have to do to calculate your cell density manually or with Hemocytap, the hemocytometer app.
Manually:
Take the average of cells per square (sum of all cells in each small square you have counted, divided by the total number of squares you have counted), multiply it by the dilution factor (if you haven’t diluted your sample, multiply by 1) and divide by the volume (in mL) of a small square, following the equation:
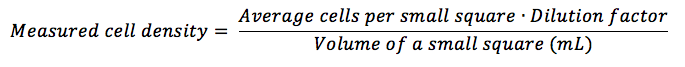
The volume of a small square is specific to the hemocytometer. It is calculated by multiplying the width by the height (which are the same – usually 1mm each) by the depth (usually 0.1mm) of a small square. In the most common case, this would be (check here to find out the volume of other squares):
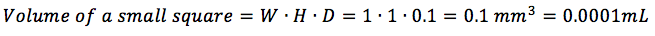
With the measured cell density obtained, you are going to calculate how much more medium you need in order to reach the manufacturer’s recommended cell density.
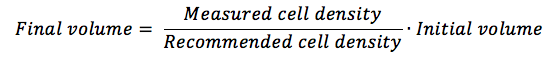
If you have already suspended the cells in some new medium, you will need to substract this from the final volume to add:
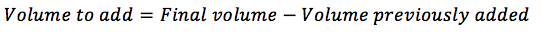
As Monsieur Malassez would say, “Voilà!”. For faster calculations, use our free hemocytometer calculator online:
Or better still, our app:
If clicking on”subculture”, introduce the dilution, target density (recommended cell density) and initial volume. Get all the calculations above done for you and read the volume you need to add. Check here for a detailed video on how to do it.
If clicking on “cell density”, introduce the dilution and the initial volume (only if you want to know the total cells). You will get the cell density (and the cell number if you gave the initial volume) as per the calculations below.
Save for your records. Ta-da!
Ho do you find what dilution is the most accurate to calculate cell concentrations for your original sample, from the density??
Thanks for your question. Please see my answer here
Hi Maria, I have a question why does the Original cell concentration (ml^-1) increase as the dilution increases ??
Hi Samuel,
The more you dilute, the less cells from the original sample remain in the diluted volume. To account for this, you multiply by the number of times you have diluted. For example, you have 100,000 cells/mL in the original sample. You dilute once (let’s say 50uL in 50uL of trypan blue; this is a 1:1 dilution or dilution factor equal to 2), the concentration should be half right? So you dilute once, the concentration in your diluted solution is 50,000 cells/mL. Now let’s say you dilute once your diluted solution: you had 50,000 cells/mL, you take 50uL from there and 50uL trypan blue or water (this is a 1:1 dilution or dilution factor equal to 2 from the diluted solution, or 1:3 dilution / dilution factor 4 from the original sample), you get 25,000 cells/mL. As you can see, in the first dilution you had a dilution factor of 2 and concentration of 50,000 cells/mL while in the second you had a dilution factor of 4 (from the original) and a concentration of 25,000 cells/mL. To calculate the original concentration backwards, you would multiply the dilution factor by the concentration. For the first dilution, this is 2 x 50,000 cells/mL = 100,000 cells/mL. For the second dilution, this is 4 x 25,000 cells/mL = 100,000 cells/mL.
Therefore, the original cell concentration is always the same for the same sample. Depending on how many times you dilute, the dilution factor will change.
Was that clear? Cheers
Maria
its very clear Thank you madam
Yes its very clear thank you 😀
Hi maria, I have a question Why some equation should to multiply by 10,000 cell/ml and multiply dilution factor?
for example this equation
[ Total cell/ml= Total cells counted x (dilution factor/# of squares) x 10,000 cell/ml ]
why they multiply by 10,000 cell/ml?
Hi there,
You multiply by the dilution factor if you want to find out the original cell concentration, i.e., previously to any dilutions you have performed specifically for counting cells. The 10,000 factor is not in cell/mL but in mL^-1 (or 1/mL). It represents the inverse of the volume of one of the corner squares, which is calculated as the area: 1 mm x 1 mm = 1 mm^2 times the height of the space between the hemocytometer and the coverslip (0.1mm), or 1 mm^2 x 0.1 mm = 0.1 mm^3 = 0.0001 mL. When you do the inverse, 1/0.0001 mL^-1 = 10,000 mL^-1 which is the factor you are using. You can find more details about these calculations in my other post on hemocytometer sizes.
Hope that helped!
Maria
thank you very much
I had the same question, I now think I understand your response above to Mr. Kiattipan and this has to do with volume of squares. I have also have noticed in your calculations for squares volume that I should use 10 000 (corner sq. 1 mm 1 mm2 0.1 mm3 0.0001 mL 4 per chamber).
I was confused seeing most people when reporting cell density, they will have average no of cells counted x dilution factor x 10 000, some would have average no of cells counted x dilution factor x 10 00000.
I counted microalgae cells in four corner squares and I believe in my case I will report cell density (x10 000 cells/mL).
Many thanks
Thank you so much !
How will you calculate the dilution for salivary Nutrophil
50ml of saliva collected,centrifuged, supernant discarded. To pellet 5ml of HBS was added. 1ml taken from and to that 20microliter of acridine orange was added. What is the dilution factor for this
Hi! so i’m trying to calculate the total amount of cells under to coverslip.
We put 20ul of blood into 5ml of saline. Therefore I calculated the dilution factor to be 251.
We counted the amount of RBC in a square at 40X on the microscope and got an average of 76 RBC.
Using the volume of 0.0001… the measured cell density is 190760000…? and also where does the recommended cell density come from?
My question is, how do I calculate the number of RBC under the coverslip AND does it matter how much of the solution I put on the slide. If so how does that work into the equation. (we put 5ul of the solution on the slide.
Sorry if that is really jumbled thoughts, im very confused.
Hi Danielle,
Glad you asked! Here’s the step-by-step of your calcs:
Did that clear up your confusion? 🙂
Maria
TLC calculation by Haemocytometer
Dear Maria
I isolated protoplast from leaves and counted it on hemocytometer, the Av. number was 111,75
i want to know how can i calculate the amount needed for concentration of (2×100000),(4×10000)
and if i had live cells av. number 20.43 and dead cells av. number 15.43 for the (4×10000) plate for example
how can i calculate viability
and the third thing
if i started with 0.22 g of fresh tissue how can i know the amount of protoplast per gram fresh tissue
waiting for your kind reply
Hi Sara,
Please see the calculations below for the amounts needed to reach those two concentrations (in here I assume a dilution and an final desired volume, just change them to the actual ones used):
For the viability:
viability = 100 x (average live cells) / (average live cells + average dead cells) = 100 x (20.42) / (20.42 + 15.43) = 56.97%
Regarding your last question, you will have to give me more information on the specific protocol you follow after the fresh tissue is processed until you get to the sample you count on the hemocytometer.
Sorry for the delay in reply!
Maria
Hi Maria,
What a great job you are doing here!
I would like to ask you: if we take into account the number of cells measured in all 25 big squares, do we still have to divide by the number of squares measured in this equation (Total cell/ml= Total cells counted x (dilution factor/# of squares) x 10,000 cell/ml) ?
For example, if I count 130 cells in all the 25 big squares (that represent 1µl?), the total number of cells would not be 130* dilution factor *10.000?
Thank you in advance
Good morning,
I’m undergrad student and I find the calculus quite complicate during my internship because I don’t know if I have to take into account the volum in which I have resuspended the cells. I explain every step that I do:
I have a T-75 flask of cells, I trypsinize them with 1,5mL of trypsin and then I add 8,5mL of medium into the flask so I can take the adherent cells and put them into a falcon. Once my cells are into the falcon I take 10uL of the sample and place it on the chamber. If I count 4 big external squares and my avarage is 74 cells, how should I proceed if I want to plate 750.000 cells/well in a 6well plate?
I’m quite desperate with knowing exactly which is the proper procedure to calculate this and any help is useful. Thank you
You can use my app, Hemocytometer Sidekick, if you want it to calculate for you. But I highly recommend understanding it yourself!
74 * 10000 (this accounts for the volume) = 740,000 cells/mL in your falcon tube. So you can tell that you’d end up adding a bit more than 1 mL of your cells in each of the wells.
(cells/well) / (cells/mL) = (mL/well)
Hi Dr.!
I want to ask about how to calculate cell/microorganisms under coverslip (without grid).
I am now study on stomach content of molluscs.
As for now, I am using 22x22mm coverslip place onto a glass slide. I have 2ml of samples. I pipette out 0.1ml of diluted samples onto the coverslip and observed under microscope.
Let say I got 3 cells/microorganisms on the total area of coverslip, do I need to times by the area of coverslip (22x22mm) and 0.1 ml of samples dropped onto the coverslip to get the total amount of cells/microorganisms in 2ml samples?
I try to used sedgewick rafter, but unfortunately I cannot used the 40x magnification as I need to identify the diatoms up to genus species. That is the reason why I have to used coverslip and glass slide to count the number of microorganisms.
I am seek for guidance on the calculation method using normal coverslip/microscope slide.
That is a great question! If you calculate that way, you might lose a bit of precision in the numbers you obtain, because it will more heavily depend on pipetting error. But if your numbers are really as low as 3 total per sample, your error will probably be dominated by stochastic variations, so maybe it would make more sense to just record counts instead of converting it into a concentration?
Can you say a bit more about why you aren’t able to use the Sedgwick-Rafter chamber? Why can’t you use the magnification you need with it?
Please I have a question. In our sector, we count budding cells separately from single cells. If you happen to count budded cells as two, must you count it again as a budded cell?
Don’t know if my question is clear. Thanks
This is an old question (apologies for the delay!) but the answer is – it very much depends! The absolute most important thing is to be consistent, and document your particular method. You might be interested in total number of units at a given time, in which case the budding cell is still a unit. But those budding cells will rapidly become two, so that concentration measurement would possibly be an underestimate; counting them as two would possibly overestimate. Maybe you’d prefer to have a concentration range instead of a single value. You might also be interested in what fraction of the population is currently budding, which is possibly why you might want separate counts.
But to reiterate the most important part: document the method and be consistent.